Lab Report on synthesis of the bidentate ligand bis(pyrazolyl)methane (bpm) and its substitution reaction with molybdenum hexacarbonyl.
The lab report below was submitted as part of the coursework for
CM3291 Advanced Experiments in Organic and Inorganic Chemistry. Please do not plagiarise from it as
plagiarism might land you into trouble with your university. Do note
that my report is well-circulated online and many of my juniors have
received soft copies of it. Hence, please exercise prudence while
referring to it and, if necessary, cite this webpage.


4. Giancardo Gioia Lobbia and Flavio Bonati. Bis(pyrazolyl)methanetetracarbonyl-chromium(0), -molybdenum(0) and -tungsten(0). Journal of Organometallic Chemistry. Vol. 366, Issues 1 – 2, 25 April 1989, pg 121 – 127.
Aim
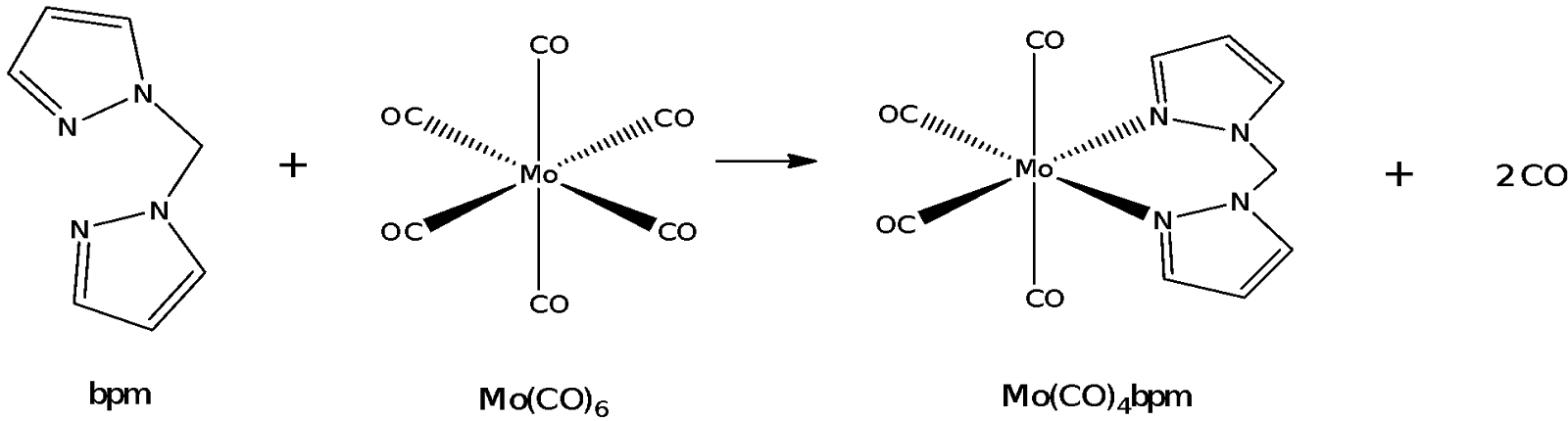
This experiment consists of two parts. The first part involves the synthesis of the bidentate ligand bis(pyrazolyl)methane (bpm). The second part of the experiment involves the bpm reacting in a ligand substitution reaction with molybdenum hexacarbonyl.
Results & Calculations
Part 1: Synthesis of the ligand (bpm)
Molar mass of pyrazole: 2 x 14.01 + 3 x 12.01 + 4 x 1.0079 = 68.08 g mol-1
Molar mass of CH2Br2: 12.01 + 2 x 1.0079 + 2 x 79.90 = 173.83 g mol-1
Molar mass of bpm: 4 x 14.01 + 7 x 12.01 + 8 x 1.0079 = 148.17 g mol-1
Mass of pyrazole: 1.0081 g
Number of moles of pyrazole: 1.0081 / 68.08 = 14.81 mmol
Volume of CH2Br2: 0.50 ml
Density of CH2Br2: 2.477 g ml-1
Mass of CH2Br2: 0.50 x 2.477 = 1.2385 g
Number of moles of CH2Br2: 1.2385 / 173.83 = 7.12 mmol
As the mole ratio of pyrazole to dibromomethane is 2:1, twice of 7.12 mmol is 14.24 mmol which is less than the number of moles of pyrazole. Thus, this means that dibromomethane was the limiting reagent in the reaction.
Mole ration of CH2Br2 to bpm is 1:1.
Number of moles of bpm: 7.12 mmol
Theoretical mass of bpm: 7.12 x 10-3 x 148.17 = 1.0550 g
Actual mass of bpm: 0.6330 g
Percentage yield of bpm: 0.6330 / 1.0550 = 60.0%
Melting point of bpm: 106.20 – 109.30 oC
Part 2: Reaction with Mo(CO)6
Molar mass of Mo(CO)6: 95.94 + 6 x (12.01 + 16.00) = 264.00 g mol-1
Molar mass of Mo(CO)4bpm: 95.94 + 4 x (12.01 + 16.00) + 148.17 = 356.15 g mol-1
Mass of bpm: 0.1508 g
Number of moles of bpm: 0.1508 / 148.17 = 1.018 mmol
Mass of Mo(CO)6: 0.2712 g
Number of moles of Mo(CO)6: 0.2712 / 264.00 = 1.027 mmol
The mole ratio of bpm to Mo(CO)6 is 1:1, and as the number of moles of Mo(CO)6 is more than that of bpm, thus bpm is the limiting reagent.
Number of moles of Mo(CO)4bpm: 1.027 mmol
Theoretical mass of Mo(CO)4bpm: 1.027 x 10-3 x 356.15 = 0.3658 g
Actual mass of Mo(CO)4bpm: 0.2196 g
Percentage yield of Mo(CO)4bpm: 0.2196 / 0.3658 = 60.0%
Temperature at which Mo(CO)4bpm decomposes: 138.70 0C
IR Spectrum Analysis
The IR spectrums of bpm and Mo(CO)4bpm were obtained using KBr disks placed in an infrared spectrometer. The following tables show the results of the IR spectrums:
Table 1: IR analysis of bpm
Wavenumber / cm-1
|
Vibrational Mode
|
Intensity
|
3134.7, 3104.3, 3017.0
|
sp2 C-H stretch
|
Weak – medium
|
2971.5
|
sp3 C-H stretch
|
Weak – medium
|
1739.9
|
Ethyl acetate C=O stretch
|
Medium
|
1623.7, 1435.8
|
C=C stretch
|
Weak, strong
|
1519.3
|
C=N stretch
|
Strong
|
1458.8, 1435.8
|
CH2, CH3 bending
|
Weak, medium
|
1391.9, 1372.0
|
sp2 C-N stretch
|
Strong
|
1204.8, 1094.4
|
Ethyl acetate C-O stretch
|
Strong
|
1291.8, 1273.8
|
sp3 C-N stretch
|
Strong
|
765.0, 734.8
|
sp2 C-H OOP bending
|
Strong
|
Table 2: IR analysis of Mo(CO)4bpm
Wavenumber / cm-1
|
Vibrational Mode
|
Intensity
|
3139.7
|
sp2 C-H stretch
|
Medium
|
3036.0, 2959.1
|
sp3 C-H stretch
|
Weak
|
2017.9, 1929.4, 1871.9, 1805.1
|
4 C≡O stretches (2 sym, 2 asym)
|
Strong
|
1516.3
|
C=N stretch
|
Medium
|
1458.6, 1428.3
|
CH2, CH3 bending
|
Medium
|
1401.6
|
sp2 C-N stretch
|
Medium
|
1283.6
|
sp3 C-N stretch
|
Strong
|
763.1
|
sp2 C-H OOP bending
|
Strong
|
1H NMR Analysis
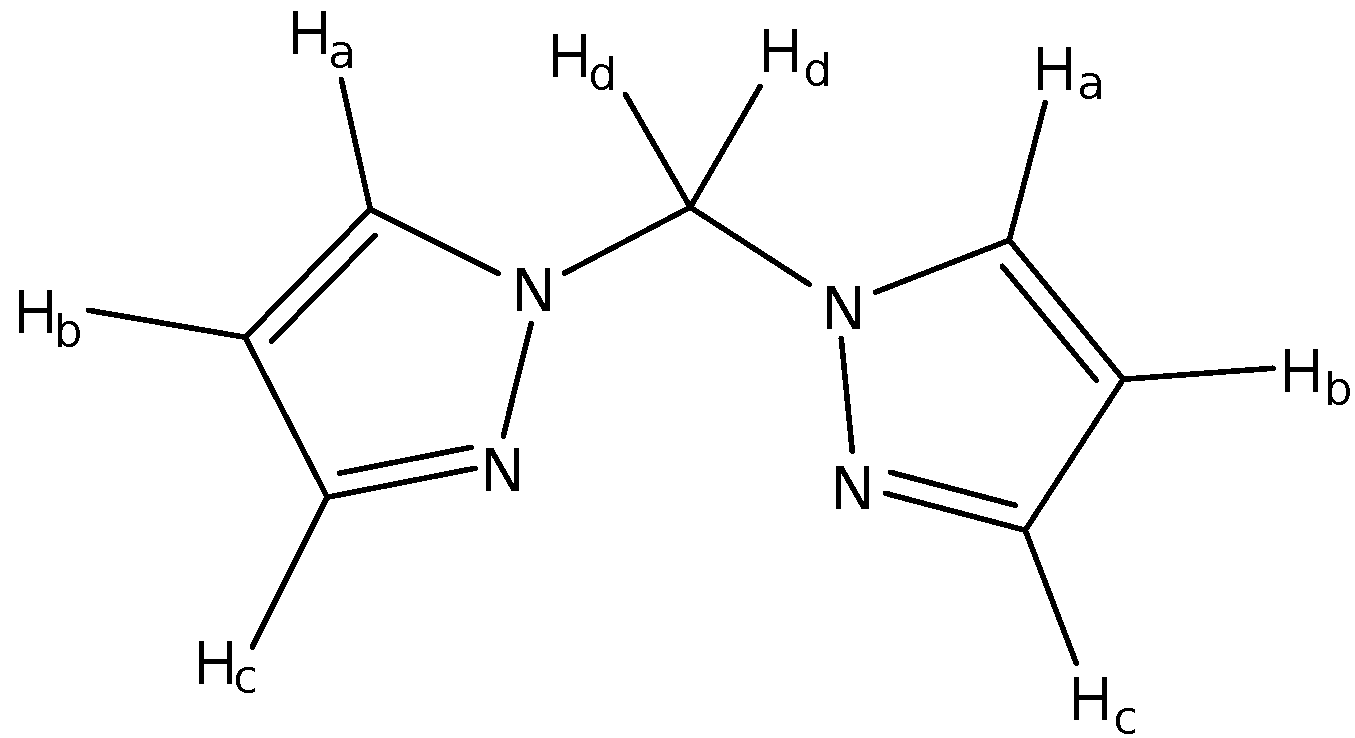
Table 3: 1H NMR Analysis of bpm
 * * |
Chemical Shift / ppm
|
Multiplicity
|
Integration
|
No. of Protons
|
Assignment
|
7.671
|
Doublet
|
1.00
|
2
|
Ha
| |
7.551
|
Doublet
|
1.03
|
2
|
Hc
| |
6.317
|
Singlet
|
1.25
|
2
|
Hd
| |
6.292
|
Triplet
|
1.01
|
2
|
Hb
| |
7.277
|
Singlet
|
-
|
-
|
CHCl3
|
Discussion
Part 1: Synthesis of the ligand (bpm)
Mechanism and Reagents used
In the first part of the experiment, the bis(pyrazolyl)methane or bpm for short, was synthesized using two equivalents of pyrazole and one equivalent of CH2Br2 in a nucleophilic substitution reaction as shown by the above mechanism. In the first step, the pyrazole is deprotonated by the hydroxide ion to form a negatively charged pyrazole. The phase-transfer catalyst Bu4N+Br- was used in the reaction to facilitate the movement of the hydroxide ion from the aqueous phase of NaOH to the organic phase where pyrazole was present. The phase-transfer catalyst is soluble in both the aqueous and organic phases. The aqueous phase is made up of the 16 ml 8M NaOH while the organic phase comprises the 30 ml of toluene. The quaternary ammonium ion on Bu4N+ is charged and can interact electrostatically with the polar molecules and ions in the aqueous phase, hence it is soluble there. The long butyl chains attached to the ammonium ion can interact with through van der Waal’s forces with the non-polar toluene molecules, and hence Bu4N+ is also soluble in the organic phase.
The presence of the phase-transfer catalyst was vital in this reaction for two reasons. Firstly, it allows the hydroxide ion to move from the aqueous layer to the organic layer where it can interact with the pyrazole molecule. This ion movement occurs when there is electrostatic attraction of Bu4N+ and OH- ions in the aqueous phase, after which the Bu4N+ OH- formed then moves into the organic layer via the organic-soluble butyl chains on the ammonium ion. Secondly, a phase-transfer catalyst is required because it prevents the flooding of hydroxide ions into the organic layer whereupon the hydroxide ion would interact with the CH2Br2 instead of pyrazolyl anion in an SN2 reaction. This is because both OH- and the pyrazolyl anion are nucleophiles which can undergo SN2 reaction with CH2Br2. With the phase-transfer catalyst, only a small amount of OH- ions are present in the organic phase at any point in time, and hence, a chemoselective reaction takes place. Deprotonation of the pyrazole molecule occurs more readily as compared to the OH- SN2 reaction with the CH2Br2.
The negatively-charged nitrogen on the pyrazolyl anion attacks the CH2Br2 in an SN2 reaction which causes the Br- to leave. An SN2 reaction is a nucleophilic substitution reaction whereby the nucleophile attacks the electrophilic carbon on one side and kicks out the leaving group on the opposite side of the carbon. As dibromomethane has two Br atoms, it is possible to have another subsequent SN2 reaction using another equivalent of the pyrazoyl anion which gives the product bis(pyrazolyl)methane. Reflux was done for 1 hr in an oil bath to speed up the reaction. After the reflux, separation was done giving the organic layer containing most of the product and the aqueous layer. The organic layer comprising toluene was less dense at 0.867 g ml-1 as compared to the aqueous layer made up of water, which has a density of 1.00 g ml-1.
Extraction and Column Chromatography
Extraction was subsequently done for the aqueous layer with dicloromethane to increase the yield of the product as much as possible. This yield in turn is dependent on the extraction partition coefficient and the number of times the solution was extracted with 20 ml of dichloromethane. In this extraction, the aqueous layer was formed at the bottom due to the presence of the phase-transfer catalyst, which increased the density of the aqueous layer relative to the organic layer. The organic layers containing the product were combined together and then dried with MgSO4, which is a hygroscopic powder that absorbs water readily from the organic mixture. Filtration was done to remove the hydrated MgSO4 after which the crude product was purified using a silica gel column prepared with ethyl acetate.
Column chromatography was prepared by using silica gel as the stationary phase and ethyl acetate as the mobile phase. The column was prepared by filling it with silica gel using ethyl acetate to flush it down the column. Cotton wool and sand were used to plug the column to prevent silica gel from leaking out whilst the separation was being done. The sample was poured on top of the silica gel and allowed to flow down the column, facilitating the adsorption of the phase-transfer catalyst Bu4N+ Br- to the silica gel. Ethyl acetate was then added to the column as the mobile phase, causing the separation of the different substances in the mixture as it flows down the column. As the phase-transfer catalyst is charged, it adheres more strongly to the polar silica gel and hence is retained in the column. However, the bpm is non-polar, flows through the column more easily and is eluted out. The ethyl acetate solvent in the eluent is then evaporated using the rotary evaporator to obtain the purified bpm. However, despite the purification by column chromatography, the final product obtained was still pinkish in colour due to the presence of some impurities that did not absorb to the silica gel. Solvent-pair recrytallization was done by first dissolving bpm in a minimum amount of hot ethyl acetate. Then, another solvent hexane can be added to the solution until a slight amount of the bpm precipitates out, which confirms that there is a hot saturated solution of bpm obtained. The solution can then be cooled in an ice bath to precipitate out the pure bpm as white crystals.
Comment on Yield and Melting point
The percentage yield of bpm is 60.0% while the melting point range of bpm is from 106.20 – 109.30 oC. The yield is reasonable but judging from the wide melting point range, impurities are likely to be present in the sample. The melting point range also falls below the theoretical melting point of 112.80 oC.
IR Spectrum Analysis of bpm
IR analysis was done on the bpm and tabulated in the results section of this report (shown in Table 1). As seen from the IR spectrum, there was a peak at 1739.9 cm-1 which is most likely the C=O stretch coming from traces of ethyl acetate left on the bpm compound. Further evidence was present as peaks at 1204.8 cm-1 and 1094.4 cm-1 which correspond to the C-O stretch from ethyl acetate. As there are sp2 and sp3 carbons in bpm, there are the characteristic C-H stretches observed in the IR spectrum. The alkene C=C stretch occurs in pairs at 1623.7 cm-1 and 1435.8 cm-1. Furthermore, there is also the C=N and C-N stretches occurring at 1519.3 cm-1 and 1270 – 1400 cm-1 respectively. These characteristic absorptions serve as evidence that the compound synthesized was bpm.
1H NMR Spectrum Analysis of bpm
The bpm was also further analyzed using 1H NMR spectroscopy with the assignments shown in Table 3. There is a total of eight protons in bpm which consists of four pairs of chemically equivalent protons as bpm is a symmetric molecule. The singlet at 6.317 ppm corresponds to Hd which is the methylene hydrogens that are not coupled to any neighbouring hydrogens. The nearest hydrogen to Hd is Ha which is more than three bonds away and hence, the coupling constant is too small to cause any appreciable splitting in the singlet peak. The triplet at 6.292 ppm corresponds to Hb which is coupled to Ha and Hc through 3Jbc and 3Jac coupling. Even though Ha and Hc are in chemically different environments, the difference in the coupling constants of 3Jbc and 3Jac is minimal. Hence, instead of a doublet of doublets, a triplet is formed instead.
For Ha and Hc, both of these proton signals are doublets. However, Hc is at a lower chemical shift (more shielded) than Ha. This could be understood from the delocalization effect which causes the carbon attached to Hc to carry a partial negative charge when the lone pair on one of the nitrogen atoms donates itself into the five-membered ring. It is illustrated as such:
As the carbon attached to Hc carries the partial negative charge, the electron cloud density is higher on this carbon and this increases the shielding effect on Hc. As such, Hc would have a signal of lower ppm as compared to Ha which does not experience the same effect.
The solvent CDCl3 is not entirely deuterated meaning that there are still some solvent molecules present as CHCl3. As such, the signal at 7.277 ppm is from the CHCl3 and in fact, other peaks are calibrated with respect to this signal. There were also other unassigned peaks present in the NMR spectrum which were possibly due to impurities present in the bpm.
Part 2: Reaction with Mo(CO)6
Mechanism and Reagents used
The synthesis of Mo(CO)4bpm involves the reflux of Mo(CO)6 with bpm. Molybdenum is a group 6 transition metal that can form coordinated complexes with ligands like CO and bpm. Mo forms an octahedral complex with 6 CO ligands or with 4 CO ligands and 1 bpm chelate. Bpm is a bidentate ligand in that it binds to the molybdenum metal centre at two sites. This latter reaction involves a ligand exchange or ligand substitution reaction whereby two of the CO ligands are substituted for a bpm ligand.
Mo has 6 valence electrons and in Mo(CO)6, each of the CO ligands contributes 2 electrons to make the total number of electrons in the complex 18. Complexes with 18 electrons are exchange inert in that it is difficult to substitute the CO ligand with another ligand like bpm, because the transition state would involve breaking the 18-electron rule of the metal complex. In any case, exchange reactions involving Mo(CO)6 would involve a dissociative substitution mechanism in which one of the CO ligands leaves in a step analogous to an SN1 pathway upon which the lone pair on one of the nitrogen atoms on bpm attacks the Mo metal centre to form Mo(CO)5bpm as an intermediate.
Mo(CO)6 + bpm -> Mo(CO)5 bpm + CO
With one nitrogen atom complexed to the molybdenum metal centre already, it becomes easier for the other nitrogen atom on bpm to coordinate to the molybdenum by displacing another CO ligand in a likely SN2 step.
Mo(CO)5 bpm -> Mo(CO)4 bpm + CO
This gives rise to a cis-conformation in which the bpm is bound to the Mo metal centre through two adjacent binding sites. Dissociative substitution reactions like in this case are entropically favourable as there is an overall increase in the total number of molecules present in the system due to chelation by the bpm molecule. Despite the reaction being entropically favourable, there is still another competing effect at work in the system. This other effect is the strength of the ligand which affects how strongly the ligand binds to the Mo metal centre. Bpm could be likened to that of bipyridine which is lower than CO in the spectrochemical series. As such, it would be difficult for a weaker field ligand like bpm to displace CO, which is a stronger ligand. This leads to the first step of the ligand exchange reaction being the rate-determining step with considerable activation energy. Thus, the reaction mixture was refluxed for 2 hours to ensure that a significant portion of bpm would react with Mo(CO)6. The second step of the ligand exchange reaction occurs more readily due to the close proximity of the ligand as well as the chelation effect once both nitrogen atoms on bpm bind to Mo.
A bubbler was used in conjunction with nitrogen gas to provide an inert environment for the reaction to take place. This prevents other gases like oxygen from interfering with the reaction possibly by oxidising the CO ligands on the Mo complex. This ensures a higher yield of Mo(CO)4 bpm. Also, the nitrogen mixes with the CO produced to dilute the toxic gas being emitted in the fume hood. This is a safety precaution for this experiment.
IR Spectrum Analysis of Mo(CO)4 bpm
Mo(CO)4 bpm was analyzed using IR spectroscopy, with the assignments shown in Table 2 in the results section. From the IR spectrum, the most significant assignment would be that of the four C≡O stretches at 2017.9, 1929.4, 1871.9 and 1805.1 cm-1. This provides evidence of the presence of a cis-octahedral complex because the four CO stretches correspond to two sets of symmetrical and asymmetrical stretches. In the complex, the two CO ligands trans to each other can be treated together, giving rise to a symmetrical and asymmetrical stretch. The other two CO ligands cis to each other are also treated together to give another pair of symmetrical and asymmetrical stretches.
All four of the above vibrational stretching modes results in a change of dipole moment and hence, are IR active. Free C≡O has a stretching frequency of ~2143 cm-1. However, when a CO ligand binds to a metal, the back donation of electron density from the metal to the carbonyl * orbital decreases the bond order of C≡O and decreases the CO stretching frequency. Thus, the greater the electron density at the metal centre, the greater the degree of back donation and hence, the lower the frequency or wavenumber of the CO stretch. Mo in the complex has six valence electrons with an electronic configuration of [Kr]5s24d4. The back donation of the valence electrons to CO decreases the stretching frequency from 2143 cm-1 and hence there is a range of frequencies from ~ 2000 – 1800 cm-1.
A cis-conformation is also more favourable in terms of the conformation of the complex as the bpm ligand is unlikely to stretch itself over two axial positions and form a trans complex. The methylene group connecting the two pyrazole molecules in bpm is too short for the entire ligand to stretch itself from one axial position on the Mo metal centre to the other axial position. Doing so would cause significant steric strain and the geometry of the complex would be distorted. Hence, from the conformational perspective, it could be observed that a cis-compound would be the more favourable complex formed.
Colour of the complex
Mo(CO)6 is white in colour whereas the product Mo(CO)4 bpm is yellow-green in colour. For Mo(CO)6, the white colour of the complex is because there are no d-d transitions occurring in the visible range of electromagnetic radiation due to the high splitting energy caused by CO, which is a strong field ligand. Hence, if d-d transitions were to occur, only photons of high energy, presumably in the UV region would be absorbed. As no visible radiation is absorbed, the colour of the complex is white. Furthermore, it is unlikely that LMCT or MLCT occurs in Mo(CO)6. LMCT is unlikely because Mo is an electron rich metal and CO ligands are poor electron-donors. MLCT is possible but it does not take place in the visible region because the * orbitals of CO are much higher in energy relative to the Mo metal orbitals. Hence, MLCT would take place in the uv region presumably. However, for Mo(CO)4 bpm, it is yellow green in colour because MLCT occurs in the visible region aided by the presence of low-lying ligand orbitals that arose from bpm. Electrons from Mo thus could be donated to the bpm ligand, causing an absorption in the region ~570 nm which gives rise to the yellow-green colour observed. Bpm is a good -acceptor ligand because it has low-lying * orbitals due to the conjugation in the two pyrazole rings.
Comment on Yield and Melting point
The percentage yield of Mo(CO)4 bpm is 60.0% while the decomposition temperature is 138.70 oC. The yield might be inflated due to the presence of impurities in the starting material of bpm, which consequently led to other side products formed that have inadvertently been mixed with Mo(CO)4 bpm. The complex decomposes as it slowly changes to dark green and eventually black upon raising the temperature.
Conclusion
Bpm was synthesized from pyrazole and CH2Br2 with a yield of 60.0% and a melting point temperature range of 106.20 – 109.30 oC. IR analysis done confirms that bpm was indeed synthesized. Also, Mo(CO)4 bpm was synthesized from Mo(CO)6 and bpm with a yield of 60.0% and a decomposition temperature of
138.70 oC. IR analysis indicates the presence of a cis-complex whereby the bidentate bpm ligand binds to two adjacent sites on the Mo metal atom.
138.70 oC. IR analysis indicates the presence of a cis-complex whereby the bidentate bpm ligand binds to two adjacent sites on the Mo metal atom.
References
1. Elena Ioachim and Garry S. Hanan. Spectroscopy and electrochemistry of new 6,6′- disubstituted-4,4′-bipyrimidine molybdenum(0) and tungsten(0) tetracarbonyl complexes. Can. J. Chem. Vol. 83, 2005, pg 1114-1119.
2. Sylvia Ernst, Yvonne Kurth and Wolfgang Kaim. Correlation between solvatochromism and back-bonding in four isomeric (α-diimine)M(CO)4 complexes, M = Cr, Mo, W. Journal of Organometallic Chemistry. Vol. 302, Issue 2, 18 March 1986, pg 211 – 215.
3. Physical properties of bis(pyrazolyl)methane. Available at: http://www.chemspider.com/14986565. Last accessed 15 May 2011.
4. Giancardo Gioia Lobbia and Flavio Bonati. Bis(pyrazolyl)methanetetracarbonyl-chromium(0), -molybdenum(0) and -tungsten(0). Journal of Organometallic Chemistry. Vol. 366, Issues 1 – 2, 25 April 1989, pg 121 – 127.
Thank you; this was very helpful for my lab assignment
ReplyDeleteYou're welcome :]
DeleteWho are the authors that I should cite? Phenomenal paper deserves to be cited correctly!!
ReplyDeletePlease just cite using the website. Don't think that there's any need for an author's name in this case!
DeleteThanks :]
Thanks for the detailed experiment method!
ReplyDeleteHave you tried the same synthesis with bromoform instead of dibromomethane?
Glad that you found it useful!
DeleteNope, I haven't tried it with bromoform but I expect the reaction to take place in a similar fashion, perhaps even more quickly.
what's the role of toluene in this reaction?
ReplyDeletereplied as well!
DeleteHi, same question here, what's the role of toluene in this reaction? Is it to make bipyridine soluble in the organic phase?
ReplyDeletehi, you need to react hydroxide with pyrazole. hydroxide is hydrophilic while pyrazole is hydrophobic. hydroxide is soluble in aqueous solution while pyrazole is soluble in the organic solvent.
Deletehence, a mix of sodium hydroxide solution and toulene is used as the solvent. this is to provide a condition which is soluble for two reactants with different hydrophobicity/hydrophilicity to react together.
provox0ac-su Jordan Lee https://wakelet.com/wake/ZjXlAZcZSLM69ZKYFPY2n
ReplyDeleteacsotderi