The lab report below was submitted as part of the coursework for
CM3291 Advanced Experiments in Organic and Inorganic Chemistry. Please do not plagiarise from it as
plagiarism might land you into trouble with your university. Do note
that my report is well-circulated online and many of my juniors have
received soft copies of it. Hence, please exercise prudence while
referring to it and, if necessary, cite this webpage.









2 strong peaks corresponding to the aromatic C-H out-of-plane bending vibrations occur at 764.14 and 678.74 cm-1 is characteristic of a monosubstituted benzene ring. These absorptions are extremely intense, resulting from strong coupling with adjacent hydrogen atoms. Overtone of aromatic rings between 2000 and 1667 cm-1 can also be used to assign ring substitution patterns. However, these bands are not really clear from the spectrum and are thus useful to a small extent in this case.
Aim
To prepare 1,2,3-triphenylcyclopropenium bromide from diphenylacetylene, potassium t-butoxide, benzal chloride and hydrogen bromide before analysing the product with Intra-red (IR) and Ultraviolet-Visible (UV-Vis) spectroscopy.
Results & Calculations
Mass of diphenylacetylene: 0.6810 g
Molar mass of diphenylacetylene: 10 x 1.0079 + 14 x 12.01 = 178.22 g mol-1
No. of moles of diphenylacetylene: 0.6810 / 178.22 = 3.821 mmol
Mass of potassium tert-butoxide: 1.7120 g
Molar mass of potassium tert-butoxide: 4 x 12.01 + 9 x 1.0079 + 39.10 = 96.21 g mol-1
No. of moles of potassium tert-butoxide: 1.7120 / 96.21 = 17.794 mmol
Vol. of toluene: 40 ml
Vol. of benzal chloride: 1.00 ml
Density of benzal chloride: 1.254 g cm-3
Mass of benzal chloride: 1.00 x 1.254 = 1.254 g
Molar mass of benzal chloride: 7 x 12.01 + 6 x 1.0079 + 2 x 35.45 = 161.02 g mol-1
No. of moles of benzal chloride: 1.254 / 161.02 = 7.788 mmol
The mole ratio of diphenylacetylene to benzal chloride to potassium tert-butoxide is 1:1:2.
Thus, diphenylacetylene is the limiting reagent.
Molar mass of 1, 2, 3-triphenylcyclopropenium bromide: 21 x 12.01 + 15 x 1.0079 + 79.90 = 347.23 g mol-1
Theoretical mass of 1, 2, 3-triphenylcyclopropenium bromide: 3.821 x 10-3 x 347.23 = 1.326 g
Actual mass of 1, 2, 3-triphenylcyclopropenium bromide obtained: 0.4330 g
Percentage yield of 1, 2, 3-triphenylcyclopropenium bromide: 0.4330 / 1.326 x 100% = 32.7%
Discussion
Proposed mechanism
The first step of the proposed mechanism involves the attack of benzal chloride with strong, bulky base t-BuOK. The acidic proton attached to the carbon is removed by t-BuOK, after which the chloride leaves in the transition state to produce the phenylchlorocarbene intermediate. The proton attached to the carbon is acidic because of its proximity to two electron withdrawing Cl substituents. Also, the negative charge on the carbon atom in the transition state is stabilised through delocalisation into the neighbouring phenyl ring.
Carbenes are highly unstable and must be formed in the presence of the compound they are to further react with. Thus, benzal chloride was added to the reaction mixture in a dropwise manner; this ensures that the carbene formed reacts immediately with diphenylacetylene before more benzal chloride is added.
The proposed mechanism for the reaction: 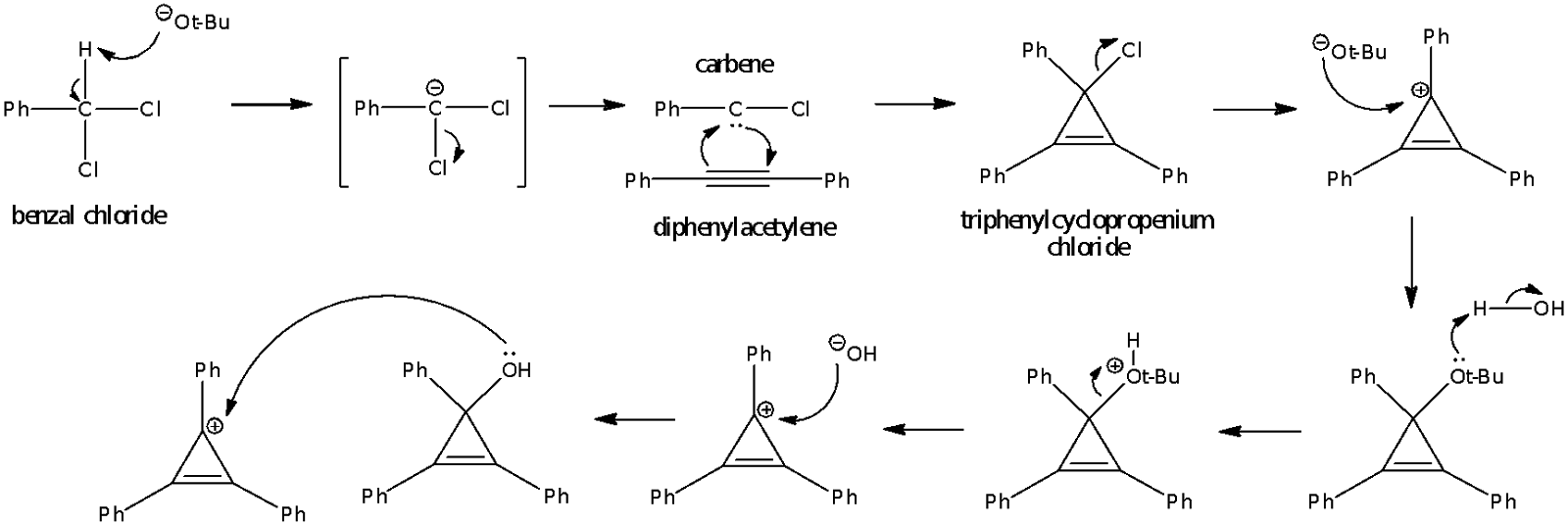
In this reaction, phenylchlorocarbene exists as the singlet carbene due to the way it was formed from benzal chloride. In the latter compound, all the electrons are paired up in 4 bonds. After the proton leaves, there are 3 bonds and an electron pair located on the carbanion. One of the chlorine atoms then leaves taking a pair of electrons from a bond with it; therefore, the remaining electrons are still paired up with two pairs in each of the C-Cl and C-Ph bonds. The remaining pair is still located on the carbene centre. This suggests that a singlet carbene is formed. The singlet carbene is stable because there is an adjacent electron-rich Cl substituent that carries lone pairs adjacent to the carbene centre.
The lone pairs of electrons on Cl are able to interact with the adjacent p orbital of the carbene which produces a new and lower energy orbital for two electrons to occupy. This further stabilizes the lone pair and encourages the electron in the p orbital to pair up with the electron in the sp2 orbital, despite electron-pair repulsion.
This molecular orbital formation moves electrons localized on chlorine into orbitals shared between carbon and chlorine and may be understood as a delocalization of the lone pair of electrons on chlorine to carbon.
Phenylchlorocarbene then reacts with diphenylacetylene to give triphenylcyclopropenium chloride in a pericyclic addition. A bond is broken and two bonds are consequently formed. This leads to a more energetically stable product as the formation of the two bonds releases sufficient energy to compensate for the energy required to break the bond.
The orientation of the phenylchlorocarbene in attacking the diphenylacetylene favors the pericyclic cycloaddition. Suppose that the phenylchlorocarbene attacks the diphenylacetylene head-on, either through the interaction of the lone pair of the carbene in its filled sp2 orbital (HOMO) with the * antibonding orbital of the acetylene (HOMO) or the empty p orbital of the carbene (LUMO) with the bond (HOMO) of the acetylene. From the illustrations on the direct approach, it could be observed that there would be a bonding interaction and an anti-bonding interaction either way. As such, due to the anti-bonding interaction, two new bonds would not be formed in this manner. Hence, another approach, whereby the carbene approaches the acetylene sideways instead, should be examined
In the sideways approach, the orientation of the orbitals would only lead to bonding interactions, thereby favoring the formation of two bonds from the breaking of a bond.
As the new bonds are being formed, the phenyl group and the chlorine atom on the carbene bends away to give a final tetrahedral arrangement about its carbon atom. This eventually gives the triphenylcyclopropenium chloride. The rearrangement of the groups attached to the carbene carbon is illustrated in the below diagram.
The next step as shown in the proposed mechanism on page 1 involves the chlorine leaving to form a planar carbocation, via an SN1 reaction. After which, there is a subsequent nucleophilic attack by the t-BuO- ion.
An SN1 reaction is preferred over an SN2 reaction for Cl- is a good leaving group and the resulting carbocation that is formed can delocalize its positive charge into the cyclopropene ring and adjacent phenyl groups. Furthermore, an SN1 reaction is favoured over an SN2 reaction because nucleophilic attack on the carbene carbon is hindered by the bulky Ph substituents. The bulky t-BuO- ion is unlikely to attack a sp3 carbon as it would form a sterically congested – therefore, unstable – intermediate. However, if it were to proceed via SN1 mechanism, the bulky nucleophile t-BuO- is free to attack on either side of the planar sp2 carbocation that forms after Cl- leaves. This also leads to the formation of triphenylcyclopropenium t-butoxide.
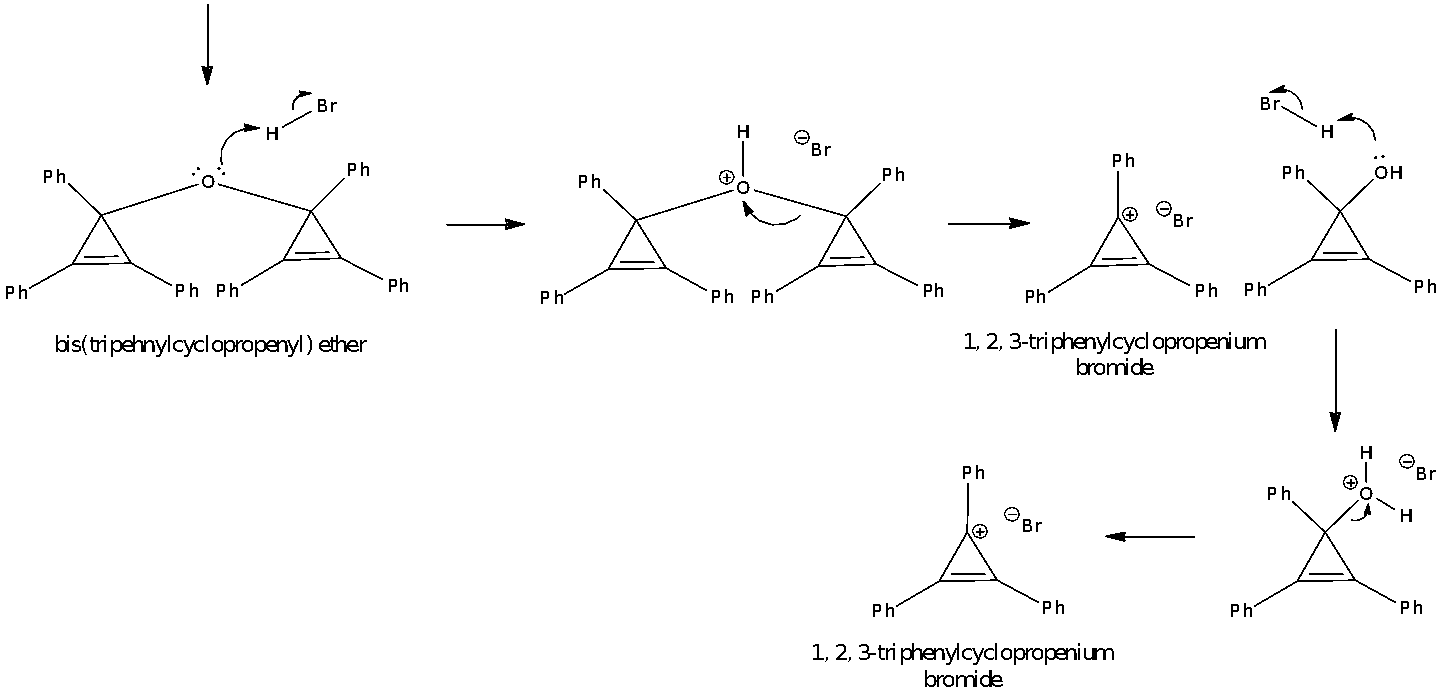
After the 3-hr reflux, upon the addition of water, the triphenylcyclopropenium t-butoxide is hydrolysed and protonated to form t-BuOH, a good leaving group. The t-BuOH formed by protonation is less nucleophilic as compared to the OH- ions which are loosely held in an ionic sphere consisting of positively charged potassium ions. Thus, it is likely for the OH- ions to attack the triphenylcyclopropenium cation in the second step of an SN1 reaction to give triphenylcyclopropenium hydroxide. The triphenylcyclopropenium hydroxide then interacts with another molecule of the cation to form bis(triphenylcyclopropenyl) ether, which dissolves in the organic solvent of toluene. Further extraction was done with diethyl ether to remove any other traces of bis(triphenylcyclopropenyl) ether that may remain in the aqueous layer.
Addition of HBr to the combined organic layers forms the 1, 2, 3-triphenylcyclopropenium bromide precipitate. In this reaction, the ether is first protonated by HBr (32% in acetic acid) which behaves as a strong acid. The protonated ether then dissociates to give triphenylcyclopropenium hydroxide and the targeted product. The hydroxide group on triphenylcyclopropenium hydroxide can also be protonated by HBr to give the product after a water molecule is lost.
Reaction Conditions
Benzal chloride was added dropwise to prevent the formation of side products and favor the intended deprotonation. Side reactions – such as an SN2 attack by deprotonated benzal chloride on unreacted benzal chloride molecules – may occur if all of benzal chloride is added at an instant, leading to a decrease in product yield.
Nitrogen gas was pumped into the system to create an inert environment. Water vapour, if present, may cause the hydrolysis of benzal chloride to benzaldehyde thereby reducing the yield of the final product.
Potassium t-butoxide was used as the non-nucleophilic base for it only deprotonates the benzal chloride in the elimination step to form phenylchlorocarbene. The bulky tertiary butyl group minimises nucleophilic attack on benzal chloride due to steric hindrance; this is especially so for benzal chloride has a bulky phenyl group next to the electrophilic carbon atom.
The addition of water purified the product by dissolving and removing inorganic salts like t-BuOH and KCl. The organic toluene layer forms the top layer in the separatory funnel as toluene is less dense (density = 0.867 g cm-3) than water (density = 1.00 g cm-3). The aqueous layer was drained into a conical flask while the organic layer was placed in a separate conical flask. The aqueous layer was then extracted twice with the mildly polar diethyl ether, which can extract the bis(triphenylcyclopropenyl) ether remaining in the aqueous layer. The organic layers were combined and dried with MgSO4, a hygroscopic powder that absorbs water. MgSO4 was then filtered off so that it would not mix with the precipitate that forms after the addition of HBr.
Huckel’s Rule
Huckel’s rule states that planar, fully conjugated, monocyclic systems with (4n + 2) electrons have a closed shell of electrons all in bonding orbitals and are stable due to their aromaticity. For the cation of 1, 2, 3-triphenylcyclopropenium bromide, there is 2 electrons and hence it obeys Huckel’s rule whereby n = 0. Thus, the cyclopropenium cation is aromatic. In addition, the positive charge can delocalize over the three attached phenyl substituents to extend the conjugated system, thereby stabilising the cation.
Analysis of IR Spectrum
The product was measured using IR spectroscopy and the assignments of the bands are as shown in the table.
Wavenumber / cm-1
|
Vibrational Mode
|
Intensity
|
3389.2
|
O-H stretch (from H2O)
|
Weak and broad
|
1594.5, 1499.4
|
Aromatic C=C stretch
|
Medium and sharp
|
1420.2
|
C=C stretch
|
Strong
|
764.0, 678.5
|
OOP C-H bend
|
Medium
|
The broad peak at 3389.2 cm-1 is due to the O-H stretch of water, which could come from HBr solution that was 32 % in acetic acid. This broad O-H stretch obscure the peaks corresponding to aromatic C-H stretch of the phenyl groups in 1,2,3-triphenylcyclopropenium bromide, which occur at 3150-3050 cm-1. Hence, sp2 C-H stretch peaks were not observed.
Aromatic C=C stretching bands occur in pairs at 1594.5 and 1499.4 cm-1. The strong absorption at 1420.2 cm-1 is due to the C=C stretching of cyclopropenium ion and this wavenumber is much lesser than that of the typical C=C stretch which occurs at around 1600 cm-1, with cyclopropene having a typical absorption around 1640 cm-1. The absorbance at a lower-than-expected wavenumber is due to the delocalisation of the double bond in the cyclopropenium ion throughout the whole compound to the three attached phenyl rings. There is some loss of double bond character for the cyclopropenium C=C, therefore leading to a corresponding decrease in its stretching frequency.
2 strong peaks corresponding to the aromatic C-H out-of-plane bending vibrations occur at 764.14 and 678.74 cm-1 is characteristic of a monosubstituted benzene ring. These absorptions are extremely intense, resulting from strong coupling with adjacent hydrogen atoms. Overtone of aromatic rings between 2000 and 1667 cm-1 can also be used to assign ring substitution patterns. However, these bands are not really clear from the spectrum and are thus useful to a small extent in this case.
The absence of strong peak at 1300-1000 cm-1 implies that ether is absent and further suggests the absence of tert-butyl ether and bis(triphenylcyclopropenyl) ether. The absence of peaks around 2250 cm-1 indicates the absence of the starting material, diphenylacetylene. Therefore, the product obtained is relatively pure.
Analysis of UV-Vis Spectrum
The solution for UV-vis measurement was prepared by dissolving 0.0120 g of 1, 2, 3-triphenylcyclopropenium bromide in 100 ml of ethanol in a 100-ml volumetric flask. This diluted solution was used to test for UV-Vis absorption.
Wavelength, (nm)
|
Absorbance, A
|
Band
|
317.20
|
1.1069
|
B-band
|
301.40
|
1.3755
|
B-band
|
288.40
|
1.1477
|
B-band
|
230.60
|
1.3533
|
K-band
|
224.20
|
1.4087
|
K-band
|
207.40
|
1.3031
|
E-band
|
Due to the extended conjugated system of the final product, it is able to absorb in the UV-Vis region. From theory, benzene has three allowed transitions which comprise the E-band at 184 nm, the K band at 204 nm and the B-band at 256 nm. These transitions arise from the HOMO-LUMO ɹ * transitions whereby the E-band is allowed while the K-band and the B-band are both forbidden transitions. These bands have been assigned to the UV-Vis spectrum results.
From the table, it could be observed that for the B-band, K-band and E-band transitions, there is an increase in the corresponding wavelength associated with the transitions. This bathochromic shift (longer wavelength absorbed) and hyperchromic effect (increase in intensity) is due to the more extensive conjugation as a result of the greater number of orbitals present in the 1,2,3- triphenylcyclopropenium ion as compared to benzene. This results in the decrease of the and * HOMO-LUMO energy gap and hence, the transitions occur with the absorption of photons of lower energy (higher wavelength).
Yield of the product
The yield of the product is 32.7% which is lower than the reported literature yield of 40%. This may be due to the loss of reagents during transfers between steps. The uneven brown colour of the final product suggests that an additional purification step might be required. Such a step might involve recrystallization of the compound whereby the impurities are left behind in the solution.
Despite all this, from absence of reactant peaks in the IR spectrum, the product obtained was rather pure. The band due to water can be removed by subjecting the synthesised product to further drying.
Conclusion
1, 2, 3-triphenylcyclopropenium bromide was synthesized from diphenylacetylene, benzal chloride, potassium t-butoxide and hydrogen bromide with a percentage yield of 32.7%. The compound synthesized was subsequently analysed with IR and UV-Vis spectroscopy to confirm its identity.
References
1. Clayden, Greeves, Warren and Wothers. Organic Chemistry. Oxford University Press, 2006. p177-177, 1058, 1060-1066.
2. Ronald Breslow & Chin Yuan, The sym-Triphenylcyclopropenyl Cation, a Novel Aromatic System , J. Am. Chem. Soc., 1958, 80 (22), pp 5991–5994.
3. F. A. Carroll. Perspectives on Structure and Mechanism in Organic Chemistry. Wiley publishers, 2nd Edition. p212-213.
4. Organic syntheses prep, http://www.orgsyn.org/orgsyn/prep.asp?prep=cv9p0730, accessed on 10 May 2012
Comments
Post a Comment